
Nicotinamide adenine dinucleotide (NAD+) is a coenzyme and considered an essential cofactor in cellular bioenergetics and adaptive stress responses. It is present in all living cells and governs fundamental biological processes including energy production, DNA repair, gene expression, calcium-dependent secondary messenger signaling and also in immune-regulatory roles. NAD+ depletion has been a subject of intense research due to the reason that it is associated with hallmarks of aging and age-related diseases, such as metabolic disorders, cancer and neurodegenerative diseases. Recent studies have suggested that physiological and pharmacological interventions that elevate cellular NAD+ levels may slow or even reverse the aspects of aging and also delay the progression of age-related diseases. In this min-review, we have described the roles of NAD+ in relationships to aging and major age-related diseases. The emphasis is on the contribution of NAD+ depletion to aging along with strategies to modulate NAD+ metabolism through physiological and pharmacological pathways. Recent human clinical studies on NAD+ boosting are summarized. We have specifically addressed how boosting NAD+ levels could potentially play an important role as a promising therapeutic strategy to counter aging-associated pathologies and accelerated aging. Finally, a brief perspective on the future research direction is presented.
Keywords: NAD+; Aging; Therapeutics; Coenzyme; Life-Extension
*E-mail: shyabiswas@gmail.com; shyabiswas@biotechkiosk.com
To cite this article: Biswas S, The Role of NAD+ in Rejuvenating Human Body, Biotechnol. kiosk, Vol 2, Issue 12, PP: 5-20 (2020); DOI: https://doi.org/10.37756/bk.20.2.12.1
- Introduction
NAD+ is recognized as an important coenzyme and cofactor in all living cells that are involved in fundamental biological processes including metabolism, cell signaling, gene expression, DNA repair, among others [1]. In earlier studies, researchers identified important role of NAD+ as a nucleoside sugar phosphate in redox reactions [2]. Recent studies have shown evidence of numerous roles of NAD+ metabolism on aging and longevity. Much focus has been placed on the age-related decline in NAD+ levels. This decline has consistently been reported as a possible consequence of an imbalance in the synthesis and consumption of NAD+ [3]. Further, decreased levels of NAD+ are considered to be associated with the hallmarks of aging and also several age-related diseases [1]. To overcome this adverse effect on aging, researchers have demonstrated a viable strategy of replenishment of NAD+ levels by administering its precursors. This route has been shown to produce beneficial effects against aging and age-related diseases. To this end, several studies based on various laboratory animal models have shown the possibility of extending lifespan of worms, flies, and rodents by boosting NAD+ levels [4-6].
With respect to the synthesis of NAD+, it is synthesized in mammals from a variety of dietary sources that include NAD+ itself, where it is metabolized in the gut and subsequently synthesized again in cells. In addition, NAD+ synthesis includes one or more of its major precursors that involve tryptophan (Trp), nicotinic acid (NA), nicotinamide riboside (NR), nicotinamide mononucleotide (NMN), and nicotinamide (NAM) [1]. In general, researchers have suggested three pathways for the synthesis of NAD+ in cells based upon the bioavailability of its precursors. These pathways are (i) from Trp by the de-novo biosynthesis pathway or kynurenine pathway (ii) from NA in the Preiss–Handler pathway and (iii) from NAM, NR, and NMN in the salvage pathway [1, 2, 7].
The role of NAD+ as a cofactor in various biological processes is vital [1]. This includes its role in the mitochondria, cytoplasm, and nucleus that drives many cellular metabolism pathways involving important processes such as glycolysis, fatty acid β-oxidation, and the tricarboxylic acid cycle. The reduced form of NAD+ is known as NADH, which is a primary hybrid donor that is used in the production of ATP via anaerobic glycolysis and mitochondrial oxidative phosphorylation (OXPHOS) [1, 8]. Further, it has been shown that NAD+ gets consumed by the NAD+-dependent sirtuins and poly (ADP-ribose) polymerases (PARPs) in the processes of protein deacetylation and poly-ADP-ribosylation (PARylation). Studies have suggested the influencing role of NAD+ on the activity of the sirtuins, which belong to a family of NAD+‐dependent deacylases that are implicated in the regulation of metabolism and mitochondrial function [9-11]. In addition to sirtuins, some other enzymes including poly ADP‐ribose polymerase (PARP) protein family and the cyclic ADP‐ribose (cADPR) synthases, such as CD38 and CD157 have been studied that have shown these enzymes or proteins to require NAD+ as a co-substrate to perform their function. The important observation of the dependence of these important metabolic enzymes including sirtuins on NAD+ levels pave the way to develop new therapeutic strategies that allow modulating their activity to achieve health benefits. This has resulted in a rapid growth of research interests in NAD+ metabolism to control and regulate aging and age-related diseases [1, 6].
Research evidence that has accumulated so far has clearly demonstrated the importance of an age-dependent decline in NAD+ levels along with an expanded role of NAD+ that includes from being a key element in intermediate metabolism to a critical regulator of multiple cell signaling pathways for beneficial therapeutic actions [12-14]. Especially, a significant body of research has focused on exploring the therapeutic potential of NAD+ boosting techniques to activate the sirtuins in a large spectrum of preclinical disease models. These models have been chosen to mimic rare genetic disorders, such as the Cockayne syndrome, as well as pandemic‐like contemporary diseases including obesity or non‐alcoholic fatty liver disease [15].
Here, we summarize the contribution of NAD+ depletion to aging and age-related diseases. We also describe potential therapeutic strategy based on boosting endogenous NAD+ levels that might be useful to counter aging-associated pathologies and/or accelerated aging.
- Age-Dependent Decline in NAD+Levels: Adverse Effects on Health
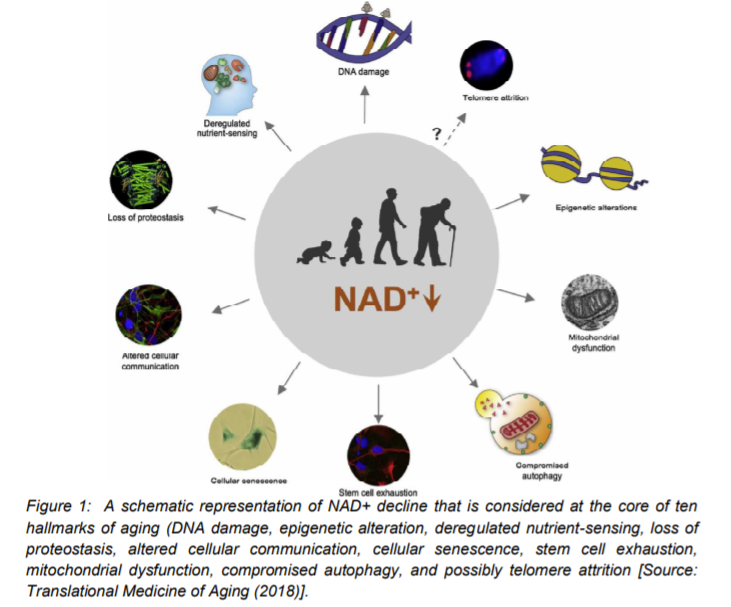
Studies have suggested that a decline in NAD+ during aging can have adverse effects on health. A decline in the NAD+ levels is believed to be a major cause of disease and disability in humans. The ongoing research has indicated that the decline in NAD+ could potentially bring hearing and vision loss, and also cognitive and motor dysfunction. This also includes immune deficiencies, autoimmunity, and dysregulation of the inflammatory response that leads to arthritis, metabolic dysfunction, and cardiovascular disease (Figure 1) [1, 16].
Total NAD+ levels were earlier thought to be highly stable. However, this previous theory of stable NAD+ levels is contradicted by the recent studies that show a steady decline in total NAD+ levels over time, which is considered to be a natural part of life for all species ranging from yeast to humans. There is considerable evidence that one of the major reasons of aging in organisms including humans is the decline in NAD+ levels as well as decreased activity of NAD+ signaling proteins [17-22].
As we discussed in the beginning, NAD+ can directly and indirectly influence many key cellular functions, including metabolic pathways, DNA repair, chromatin remodeling, cellular senescence and immune cell function. These cellular processes and functions are critical for maintaining tissue and metabolic homeostasis and for healthy ageing. Past and recent studies have repeatedly shown a gradual decline in tissue and cellular NAD+ levels in multiple model organisms that include rodents and humans. This decline in NAD+ levels has been linked with number of age-related diseases, such as cognitive decline, cancer, metabolic disease, sarcopenia, and frailty [23]. Many studies have suggested the possibility of slowing down and even reversing of many of these ageing-associated diseases by restoring NAD+ levels. Therefore, a top current research focus is to explore targeting NAD+ metabolism as a potential therapeutic approach. This is aimed at developing strategies for ameliorating age-related disease that could be leveraged to extend the healthy lifespan. Ongoing studies are directed to understand primarily the influence of NAD+ on human health and ageing biology. It is believed that a better understanding of the molecular mechanisms that regulate NAD+ levels could lead to a therapeutic pathway to effectively restore NAD+ levels during ageing. In addition, the focus is also on the issues related to the safety and the beneficial effects of the use of NAD+ repletion in ageing humans [23].
3. Modulation of NAD+ Metabolism:
Physiological and Pharmacological Strategies Research advances made so far in NAD+ metabolism and aging have indicated that therapeutics that target human aging could ultimately lead to desired healthy aging and the improvement of the quality of life. The growing body of research evidence suggests that NAD+ boosters can potentially have profound effects on the health and survival of mammals by raising NAD+ level. Therefore, inhibiting the age-related decline in NAD+ levels is considered to be critical for slowing or reversing age- or disease-related frailties [16, 24].
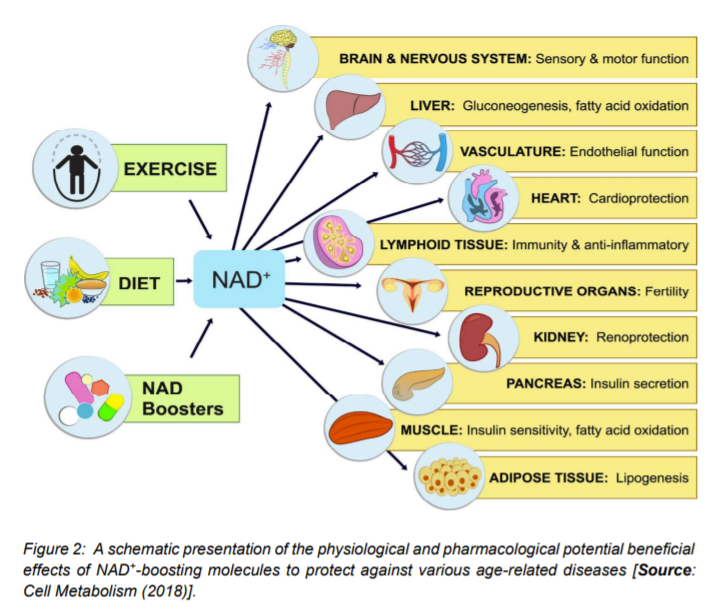
Figure 2 summarizes possible physiological and pharmacological ways that could be employed for the beneficial effects of NAD+ for improving the quality of life. This could be achieved by blocking or delaying numerous pathological hallmarks of aging that could lead to delaying age-related diseases [16]. With respect to exploring physiological strategies, it has been suggested that exercise, fasting, and maintaining a healthy diet could boost NAD+ levels (Figure 2) [1, 16]. The strategies related to the pharmacologically boosting of NAD+ involve supplementation of its precursors, NR and NMN, and also inhibition of its consumers by use of CD38 and PARPs inhibitors. Several health benefits have been suggested that could potentially result from increases in NAD+ by following the physiological and pharmacological strategies. It is believed that it could promote cognitive and sensory function, gluconeogenesis in liver, lipogenesis in adipose tissue, insulin secretion in pancreas, and insulin sensitivity in muscle [16].
Some recent studies have shown the potentials of NAD+ in protecting against cardio- and cerebrovascular disease. NAD+ can regulate immune function and inflammation and protect against acute injury in kidney. By activation of sirtuins, it can also promote and extends fertility in both males and females, ostensibly (Figure 2) [10, 16]. Further, preclinical evidence of NAD+ replenishment has suggested that it could potentially delay and/or prevent metabolic conditions, hearing loss, muscle atrophy, and cognitive decline. In addition, NAD+ precursors, especially NR has been shown to be safely administrated and also able to demonstrate improvement of cardiovascular functions in human. These results imply that a possible translational aspect of preclinical benefits of NAD+ supplementation could be a promising avenue to test the impact of elevated NAD+ biosynthesis in aging and age-associated diseases in human [15, 24].
With respect to the ways to increasing NAD+ levels, researchers have shown that the levels can be increased either by promoting its synthesis or by enhancing the enzymes that are involved in NAD+ biosynthesis or administration of NAD+ precursor molecules. This can also be achieved by limiting its consumption. It has been shown that supplementation with NA, NAM, NR, NMN, or tryptophan can increase NAD+ content [5]. Other routes have been shown that include overexpressing or activating enzymes catalyzing the rate‐limiting steps of NAD+ biosynthesis to boost NAD+ levels [25-29].
With respect to pharmacological or genetic inhibition of non‐sirtuin NAD+ consumers, such as PARP‐1 or CD38, it has been shown that such inhibition can help to preserve NAD+ levels for sirtuin activation. This is especially true in situations when non‐sirtuin NAD+ consumers are over activated. This can be understood from a typical DNA damage, which can cause a dramatic decline in NAD+ intracellular levels. This is due to PARP activation and overexpression of CD38 in cells that has been shown to result in a ~35% decrease in NAD+ levels. However, in sharp contrast, Cd38−/− and PARP‐1−/− in mice have been shown to increase NAD+ content in different organs [30-35].
In obesity and type 2 diabetes, researchers reported an increased level of nicotinamide methyl transferase (NNMT) expression. NNMT is known as the enzyme, which catalyzes the transformation of NAM into methylnicotinamide (MNA). NNMT has been shown to be highly expressed in liver and adipose tissue. Researchers have studied the possibility of inhibiting NNMT in these tissues to increase NAD+ content due to the fact that NAM would not be degraded but exclusively reconverted into NAD+. To this end, it was shown that the knockdown of NNMT resulted in increased NAD+ levels in adipose tissue. However, that was not found in the liver [15, 36, 37]. We will describe some of these therapeutic targets in the following section.
4. Therapeutic Approaches: Inhibition of NAD+ Degradation
Several studies have suggested that NAD+ levels can be boosted by direct activation of NAD+ biosynthetic enzymes. Especially, those enzymes are considerable favorable that catalyze the rate-limiting steps of de novo synthesis and salvage pathways. For example, researchers have shown that the NAM salvage pathway is the predominant route in mammalian NAD+ biosynthesis. It has been shown in this process that NAMPT is the rate-limiting enzyme in the conversion of NAM to NAD+ [38]. While, NAMPT activity can be sustained under normal conditions to maintain NAD+ homeostasis, it has been observed that its activity declines with age occurs and especially it gets exacerbated by acute lung injury, atherosclerosis, cancer, diabetes, rheumatoid arthritis, and sepsis. Researchers have shown an attractive therapeutic approach based on increasing systemic NAD+ biosynthesis with small chemical NAMPT activators. To this end, researchers identified several NAMPT-activating compounds by using a fluorometric NAMPT activity assay [39]. With respect to a neuroprotective agent, P7C3, some studies have suggested that it may be a relatively weak NAMPT activator in-vitro. Overall, NMNATs have been suggested as attractive therapeutic targets for raising NAD+ in cells. The fact is that they offer dual substrate specificity for NMN and nicotinic acid mononucleotide (NaMN) along with their ability to contribute to both de novo and salvage pathways make them promising targets [16].
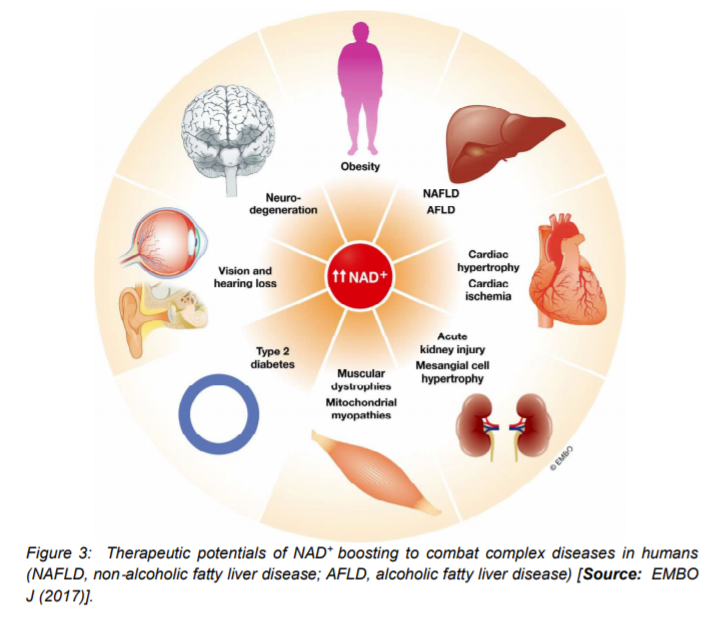
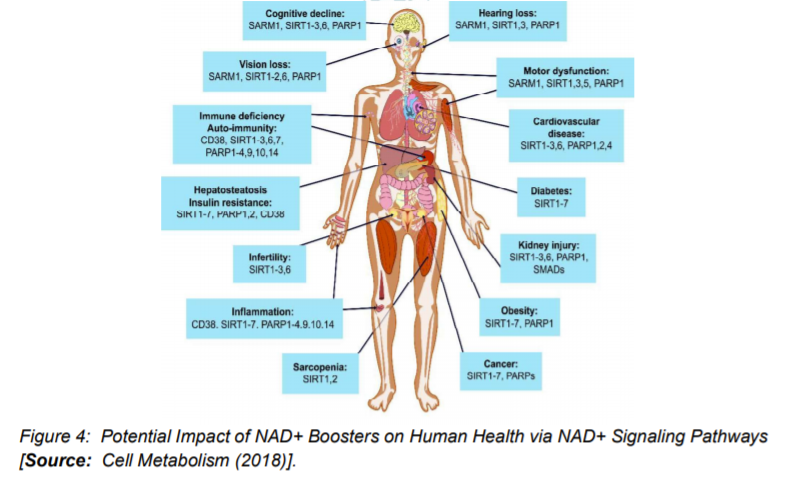
A number of animal studies have suggested that by inhibiting PARPs or NADases, also known as glycohydrolases could help boost NAD+ levels in humans to combat complex diseases (Figure 3) [15]. To this end, researchers inhibited in-vitro the major NADase in mammals, CD38 at low micromolar concentrations by using flavonoids including luteolinidin, kuromanin, luteolin, quercetin, and apigenin (IC50 < 10 μM) [40]. These molecules also appeared to target CD38 in-vivo. It has been reported that some therapeutic targets such as apigenin can increase NAD+ levels in multiple tissues that can result in decreasing global proteome acetylation. This subsequently can improve glucose and lipid homeostasis in obese mice, ostensibly by increasing activity of SIRT1 and SIRT3 (Figure 4) [16, 40]. In other reports, luteolinidin was shown to prevent the loss of NAD+ that preserved endothelial and myocardial function in the post-ischemic heart [41]. With respect to supplements, GlaxoSmithKline developed thiazoloquin(az)olinones, such as the compound 78c that showed greater potency than the flavonoids to boost NAD+ levels in plasma, liver, and muscle [42].
Despite these advances, the target specificity of 78c is still unknown and also more studies are needed to reveal whether or not it enters cells rather than acting on extracellular CD38 [16]. It is to be mentioned that in chemotherapy or monotherapies for cancer, PARP1 inhibitors are marketed as adjuncts [43]. Some studies have shown the potential of another therapeutic target, SARM1 (Figure 4), which is another NADase that initiates a local axonal degeneration after nerve injury. The pathway involves the rapid breakdown of NAD+ to ADPR, cADPR, and NAM [44]. Further, the potential of XAV939, which is a putative SARM1 inhibitor that also inhibits PARP5a (TNKS) and 5b (TNKS2) has been identified in a chemical genetic screen that can boost NAD+ levels [44]. XAV939 is known to possess excellent pharmacokinetic properties. It is currently in clinical development to treat neurological disorders and axonal injury. However, more studies are needed to gain insights into the in-vivo functions of XAV939 functions by inhibiting SARM1 or other targets [16].
- Clinical Trials of NAD+Boosters in Humans
The actions of NAD+ boosters have been studied in a number of mouse models that have shown effectiveness of NAD+ boosters to prevent or treat a variety of different diseases. These encouraging results have prompted clinical trials for NAD+ boosters in human that are safe and effective as drugs to treat both rare and common diseases and, potentially, aging itself (Figure 4) [16]. To this end, researchers investigated NA (niacin) NAD+ precursors in humans. The choice of niacin is due to the reason that in large doses (greater than a gram), administration of NA is shown to be an effective way to treat hypercholesterolemia, as it lowers LDL. Further, it is one of the few drugs that significantly raises HDL [45]. This compound is commercially available either as compressed NA (Niacor) or extended-release to prevent the flushing caused by prostaglandin release (Niaspan, Advicor, Simcor). Niaspan is used in combination with statins for the treatment of primary hyperlipidemia and mixed dyslipidemia [16]. Given the demonstrated potential of Niacore to raise NAD+ levels in rodents, current studies have focused on the possibility of NA improving cholesterol profiles in humans by raising NAD+ levels. In addition, the two compounds NA and NAM have been explored for other health benefits that include treatments for acne, kidney diseases, lupus, AD, schizophrenia, diabetes mellitus, non-small-cell lung carcinoma, obesity, HIV-induced dyslipidemia, NAFLD, sickle cell disease etc [46, 47].
A clinical trial based on a randomized, double-blind, three-arm crossover pharmacokinetic study in 12 human subjects showed that NR raised NAD+ by as much as 2.7-fold in human blood with a single oral dose of 1,000 mg. In this study, NAAD emerged as a sensitive biomarker [48]. Further, researchers conducted a clinical trial with 140 participants that were given orally administered NR. This revealed a dose-dependent increase in NAD+ from 250 to 1,000 mg/day, plateauing at a 2-fold increase in NAD+ at day 9 [49]. A similar study employing NR showed its positive effects on vascular endothelial function in healthy middle-aged and older adults. However, more investigations of motor and cognitive changes need to be done [50]. Some other studies evaluated the effects of NR on important medical parameters such as muscle mitochondrial function, cognition, immune function, kidney function, TBI, brown fat activity, lipid accumulation, energy metabolism, cardiovascular risk, body composition, and acetylcarnitine levels [16].
There have been progresses made in clinical trials at the industrial level as well, in which a pipeline of novel NAD+ precursors, for example MIB-626, have been tested [16]. Luteolin, a CD38 inhibitor was investigated that showed positive neuroprotective effects on children with autism [51]. Further, SARM1, which is a promising therapeutic target for axonopathies is the focus of preclinical development [16]. PARP1/2 inhibitors also showed improvement in the health of mice on a HFD [52, 53].
More recently, researchers conducted a pilot study and investigated changes in the human plasma and urine NAD+ metabolome using a 6 Hour intravenous infusion of NAD+ [54]. In this study, accumulated evidence suggested that active maintenance of optimal levels of the essential pyridine nucleotide, nicotinamide adenine dinucleotide (NAD+) could be beneficial in medical conditions of either increased NAD+ turnover or inadequate synthesis. These medical conditions include Alzheimer’s disease and other neurodegenerative disorders and the aging process [54]. Researchers documented changes in plasma and urine levels of NAD+ and its metabolites during and after a 6 h 3 μmol/min NAD+ intravenous (IV) infusion. They showed that the characterization of these changes could potentially help progress the development and improvement of NAD+ based treatment regimens for disorders. This clinical trial is anticipated to pave the way to potential breakthroughs in new therapeutic avenues that are likely to benefit from increased NAD+ availability including complex neurologic conditions requiring increased cellular regeneration and repair such as Alzheimer’s and other neurodegenerative dementias [54].
6. Conclusion and Perspective
We have described the role of depletion of NAD+ levels in aging in humans. Studies have shown a balanced level of NAD+ is required for maintaining proper cellular function. It has also been shown that cells can adapt different ways to regulate the biosynthesis of NAD+ that include gene regulation, feedback inhibition, compartmentalization of enzymes and intermediate metabolites. Subsequently, cells coordinate this biosynthesis via nutrient and energy sensing. The current research focus has been on boosting NAD+ levels, which is considered an important strategy to achieve the desired healthy aging and the improvement of the quality of life. We have presented and discussed important physiological and pharmacological strategies on potential routes to boost NAD+ levels. Further, there are some studies that have identified novel regulators of NAD+ homeostasis. However, more studies are required to be done to gain insights into this regulation process. One example can be considered in yeast, where NAM can replenish NAD+ pools by entering the NA/NAM salvage pathway. The other option is by de-repressing the de novo pathway via inhibiting the activity of the NAD+-dependent sirtuin Hst1 in yeast. However, it is not understood fully whether the de novo pathway in other organisms is also repressed by NAD+ and de-repressed by NAM in a sirtuin-dependent manner.
Some studies have attempted to address the repression of de novo activity by NAD+ that has been observed in bacteria. However, it has not been explicitly shown that the appearance of NAM occurs to de-repress de novo NAD+ synthesis activity in E. coli. In light of this uncertainty, future studies could target to determine whether sirtuins also play a role in the regulation of de novo NAD+ biosynthesis in higher eukaryotes, since sirtuins are highly conserved across species. Such studies are especially important as metabolites of the de novo pathway have been linked to several brain disorders. It is also anticipated that future studies would also focus on to gain a better understanding of the multiple roles of NAD+ intermediates along with novel factors that regulate NAD+ homeostasis.
Recent clinical trials in humans have suggested that NAD+ metabolism could be an emerging therapeutic target for several human diseases. To this end, it has been shown that supplementation of specific NAD+ precursors can be combined with genetic modifications and inhibitors of specific NAD+ biosynthesis steps. This can subsequently help channel the precursors to a more efficient NAD+ synthesis route. Further, several studies have reported that specific NAD+ metabolites and NAD+ biosynthesis enzymes to have additional functions. Therefore, it can be argued that studies that help understand the molecular basis and interconnection of multiple NAD+ metabolic pathways are important for the development of disease-specific therapeutic strategies. Such studies could be undertaken since these strategies have shown to be more effective if associated defects in specific NAD+ biosynthesis pathways/steps are known.
References
[1] Yahyah Aman, Yumin Qiu, Jun Tao, Evandro F.Fang, Therapeutic potential of boosting NAD+ in aging and age-related diseases, Translational Medicine of Aging, 2, 30-37(2018), DOI: https://doi.org/10.1016/j.tma.2018.08.003
[2] J. Yoshino, J.A. Baur, S.I. Imai, NAD(+) intermediates: the biology and therapeutic potential of NMN and NR, Cell Metabol., 27, 513-528 (2018), DOI: https://doi.org/10.1016/j.cmet.2017.11.002
[3] E. Verdin, NAD(+) in aging, metabolism, and neurodegeneration, Science, 350, 1208-1213 (2015), DOI: https://doi.org/10.1126/science.aac4854.
[4] G. Magni, A. Amici, M. Emanuelli, G. Orsomando, N. Raffaelli, S. Ruggieri
Enzymology of NAD+ homeostasis in man, Cell. Mol. Life Sci., 61, 19-34 (2004), DOI: https://doi.org/10.1007/s00018-003-3161-1.
[5] C. Canto, K.J. Menzies, J. Auwerx, NAD(+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus
Cell Metabol., 22, 31-53 (2015), DOI: http://dx.doi.org/10.1016/j.cmet.2015.05.023.
[6] K. Yaku, K. Okabe, T. Nakagawa, NAD metabolism: implications in aging and longevity, Ageing Res. Rev., 47, 1-17 (2018), DOI: https://doi.org/10.1016/j.arr.2018.05.006.
[7] M. Misiak, R. Vergara Greeno, B.A. Baptiste, P. Sykora, D. Liu, S. Cordonnier, et al.
DNA polymerase beta decrement triggers death of olfactory bulb cells and impairs olfaction in a mouse model of Alzheimer’s disease, Aging Cell, 16, 162-172 (2017), DOI: https://doi.org/10.1111/acel.12541.
[8] D.C. Wallace, Mitochondria and cancer, Nat. Rev. Cancer, 12, 685-698 (2012), DOI: https://doi.org/10.1038/nrc3365.
[9] Imai S, Armstrong CM, Kaeberlein M, Guarente L Transcriptional silencing and longevity protein Sir2 is an NAD‐dependent histone deacetylase. Nature 403: 795–800 (2000), DOI: https://doi.org/10.1038/35001622.
[10] Houtkooper RH, Pirinen E, Auwerx J, Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol 13: 225–238 (2012), DOI: https://doi.org/10.1038/nrm3293.
[11] Haigis MC, Sinclair DA Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol 5: 253–295 (2010), DOI: https://doi.org/10.1146/annurev.pathol.4.110807.092250
[12] Sofie Lautrup, David A. Sinclair, Mark P. Mattson, and Evandro F. Fang, NAD+ in Brain Aging and Neurodegenerative Disorders, Cell Metabolism, Cell Metabolism 30, 630-655 (2019), DOI: https://doi.org/10.1016/j.cmet.2019.09.001
[13] L.E. Navas, A. Carnero, NAD+ metabolism, stemness, the immune response, and cancer. Sig Transduct Target Ther 6, 2-20, (2021). https://doi.org/10.1038/s41392-020-00354-w
[14] Mario Romani, Vincenzo Sorrentino, Chang-Myung Oh, Hao Li, Tanes Imamura de Lima, Hongbo Zhang, Minho Shong, Johan Auwerx. NAD+ boosting reduces age-associated amyloidosis and restores mitochondrial homeostasis in muscle. Cell Reports, 2021; 34 (3): 108660 DOI: 10.1016/j.celrep.2020.108660.
[15] Elena Katsyuba, Johan Auwerx, Modulating NAD+ metabolism, from bench to bedside, EMBO J 36:2670-2683, (2017), DOI: https://doi.org/10.15252/embj.201797135
[16] Luis Rajman, Karolina Chwalek, David A.Sinclair, Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence, Cell Metabolism, 27, 529-547 (2018), DOI: https://doi.org/10.1016/j.cmet.2018.02.011.
[17] Balan V, Miller GS, Kaplun L, Balan K, Chong ZZ, Li F, Kaplun A, VanBerkum MF, Arking R, Freeman DC, et al. Life span extension and neuronal cell protection by Drosophila nicotinamidase. J Biol Chem. 283:27810–27819 (2008), DOI: https://doi.org/ 10.1074/jbc.M804681200.
[18] Belenky P, Racette FG, Bogan KL, McClure JM, Smith JS, Brenner C. Nicotinamide riboside promotes Sir2 silencing and extends lifespan via Nrk and Urh1/Pnp1/Meu1 pathways to NAD+ Cell;129:473–484 (2007), DOI: https://doi.org/10.1016/j.cell.2007.03.024. .
[19] Massudi H, Grant R, Braidy N, Guest J, Farnsworth B, Guillemin GJ. Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS One;7:e42357 (2012), DOI: https://doi.org/10.1371/journal.pone.0042357.
[20] Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Canto C, Mottis A, Jo YS, Viswanathan M, Schoonjans K, et al. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell;154:430–441 (2013), DOI: https://doi.org/10.1016/j.cell.2013.06.016.
[21] Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P, D’Amico D, Ropelle ER, Lutolf MP, Aebersold R, et al. NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science;352:1436–1443 (2016), DOI: https://doi.org/10.1126/science.aaf2693.
[22] Zhou M, Ottenberg G, Sferrazza GF, Hubbs C, Fallahi M, Rumbaugh G, Brantley AF, Lasmezas CI. Neuronal death induced by misfolded prion protein is due to NAD+ depletion and can be relieved in vitro and in vivo by NAD+ replenishment. Brain;138:992–1008 (2015), DOI: https://doi.org/10.1093/brain/awv002.
[23] A.J. Covarrubias, R. Perrone, A. Grozio, et al. NAD+ metabolism and its roles in cellular processes during ageing. Nat Rev Mol Cell Biol 22, 119–141 (2021). https://doi.org/10.1038/s41580-020-00313-x.
[24] Trevor Croft, Padmaja Venkatakrishnan and Su-Ju Lin, NAD+ Metabolism and Regulation: Lessons From Yeast, Biomolecules 10(2), 330, (2020), DOI: https://doi.org/10.3390/biom10020330
[25] Araki T, Sasaki Y, Milbrandt J, Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 305: 1010–1013 (2004); DOI: https://doi.org/ 10.1126/science.1098014.
[26] Sasaki Y, Araki T, Milbrandt J, Stimulation of nicotinamide adenine dinucleotide biosynthetic pathways delays axonal degeneration after axotomy. J Neurosci 26: 8484–8491 (2006); DOI: https://doi.org/10.1523/JNEUROSCI.2320-06.2006.
[27] Hsu CP, Oka S, Shao D, Hariharan N, Sadoshima J (2009) Nicotinamide phosphoribosyltransferase regulates cell survival through NAD+ synthesis in cardiac myocytes. Circ Res 105: 481–491 (2009); DOI: https://doi.org/10.1161/CIRCRESAHA.109.203703.
[28] Wang G, Han T, Nijhawan D, Theodoropoulos P, Naidoo J, Yadavalli S, Mirzaei H, Pieper AA, Ready JM, McKnight SL (2014a) P7C3 neuroprotective chemicals function by activating the rate‐limiting enzyme in NAD salvage. Cell 158: 1324–1334 (2014a); DOI: https://doi.org/10.1016/j.cell.2014.07.040
[29] Williams PA, Harder JM, Foxworth NE, Cochran KE, Philip VM, Porciatti V, Smithies O, John SW (2017) Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science 355: 756–760 (2017); DOI: https://doi.org/ 10.1126/science.aal0092.
[30] Aksoy P, Escande C, White TA, Thompson M, Soares S, Benech JC, Chini EN, Regulation of SIRT 1 mediated NAD dependent deacetylation: a novel role for the multifunctional enzyme CD38. Biochem Biophys Res Comm 349: 353–359 (2006a); DOI: https://doi.org/10.1016/j.bbrc.2006.08.066.
[31] Barbosa MT, Soares SM, Novak CM, Sinclair D, Levine JA, Aksoy P, Chini EN, The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet‐induced obesity. FASEB J 21: 3629–3639 (2007); DOI: https://doi.org/10.1096/fj.07-8290com.
[32] Bai P, Cantó C, Oudart H, Brunyánszki A, Cen Y, Thomas C, Yamamoto H, Huber A, Kiss B, Houtkooper RH, Schoonjans K, Schreiber V, Sauve AA, Menissier‐de Murcia J, Auwerx J, PARP‐1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab 13: 461–468 (2011); DOI: https://doi.org/10.1016/j.cmet.2011.03.004.
[33] Mukhopadhyay P, Rajesh M, Cao Z, Horváth B, Park O, Wang H, Erdelyi K, Holovac E, Wang Y, Liaudet L, Hamdaoui N, Lafdil F, Haskó G, Szabo C, Boulares AH, Gao B, Pacher P. Poly (ADP‐ribose) polymerase‐1 is a key mediator of liver inflammation and fibrosis. Hepatology 59: 1998–2009 (2014); DOI: https://doi.org/10.1002/hep.26763.
[34] Ryu D, Zhang H, Ropelle ER, Sorrentino V, Mazala DA, Mouchiroud L, Marshall PL, Campbell MD, Ali AS, Knowels GM, Bellemin S, Iyer SR, Wang X, Gariani K, Sauve AA, Canto C, Conley KE, Walter L, Lovering RM, Chin ER et al, NAD+ repletion improves muscle function in muscular dystrophy and counters global PARylation. Sci Transl Med 8: 361ra139 (2016); DOI: https://doi.org/ 10.1126/scitranslmed.aaf5504.
[35] Gariani K, Ryu D, Menzies KJ, Yi HS, Stein S, Zhang H, Perino A, Lemos V, Katsyuba E, Jha P, Vijgen S, Rubbia‐Brandt L, Kim YK, Kim JT, Kim KS, Shong M, Schoonjans K, Auwerx J, Inhibiting poly ADP‐ribosylation increases fatty acid oxidation and protects against fatty liver disease. J Hepatol 66: 132–141 (2017); DOI: https://doi.org/10.1002/hep.28245.
[36] Kraus D, Yang Q, Kong D, Banks AS, Zhang L, Rodgers JT, Pirinen E, Pulinilkunnil TC, Gong F, Wang YC, Cen Y, Sauve AA, Asara JM, Peroni OD, Monia BP, Bhanot S, Alhonen L, Puigserver P, Kahn BB, Nicotinamide N‐methyltransferase knockdown protects against diet‐induced obesity. Nature 508: 258–262 (2014); DOI: https://doi.org/10.1038/nature13198.
[37] Riederer M, Erwa W, Zimmermann R, Frank S, Zechner R, Adipose tissue as a source of nicotinamide N‐methyltransferase and homocysteine. Atherosclerosis 204: 412–417 (2009); DOI: https://doi.org/10.1016/j.atherosclerosis.2008.09.015.
[38] Revollo JR, Grimm AA, Imai S. The regulation of nicotinamide adenine dinucleotide biosynthesis by Nampt/PBEF/visfatin in mammals. Curr Opin Gastroenterol, 23:164–170 (2007a); DOI: https://doi.org/ 10.1097/MOG.0b013e32801b3c8f.
[39] Zhang RY, Qin Y, Lv XQ, Wang P, Xu TY, Zhang L, Miao CY. A fluorometric assay for high-throughput screening targeting nicotinamide phosphoribosyltransferase. Anal Biochem;412:18–25 (2011); DOI: https://doi.org/10.1016/j.ab.2010.12.035.
[40] Escande C, Nin V, Price NL, Capellini V, Gomes AP, Barbosa MT, O’Neil L, White TA, Sinclair DA, Chini EN. Flavonoid apigenin is an inhibitor of the NAD+ ase CD38: implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes;62:1084–1093 (2013); DOI: https://doi.org/10.2337/db12-1139.
[41] Boslett J, Hemann C, Zhao YJ, Lee HC, Zweier JL. Luteolinidin Protects the Postischemic Heart through CD38 Inhibition with Preservation of NAD(P)(H) J Pharmacol Exp Ther;361:99–108 (2017); DOI: https://doi.org/10.1124/jpet.116.239459.
[42] Haffner CD, Becherer JD, Boros EE, Cadilla R, Carpenter T, Cowan D, Deaton DN, Guo Y, Harrington W, Henke BR, Jeune MR, Kaldor I, Milliken N, Petrov KG, Preugschat F, Schulte C, Shearer BG, Shearer T, Smalley TL Jr, Stewart EL, Stuart JD, Ulrich JC, J Med Chem; 58(8):3548-71 (2015); DOI: https://doi.org/10.1021/jm502009h.
[43] Brown JS, Kaye SB, Yap TA. PARP inhibitors: the race is on. Br J Cancer; 114:713–715 (2016); DOI: https://doi.org/10.1038/bjc.2016.67.
[44] Gerdts J, Brace EJ, Sasaki Y, DiAntonio A, Milbrandt J. SARM1 activation triggers axon degeneration locally via NAD(+) destruction. Science, 348:453–457 (2015); DOI: https://doi.org/10.1126/science.1258366.
[45] Garg A, Sharma A, Krishnamoorthy P, Garg J, Virmani D, Sharma T, Stefanini G, Kostis JB, Mukherjee D, Sikorskaya E. Role of Niacin in Current Clinical Practice: A Systematic Review. Am J Med; 130:173–187 (2017); DOI: https://doi.org/10.1016/j.amjmed.2016.07.038. .
[46] Canto C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, Fernandez-Marcos PJ, Yamamoto H, Andreux PA, Cettour-Rose P, et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab;15:838–847 (2012); DOI: https://doi.org/10.1016/j.cmet.2012.04.022. .
[47] Gariani K, Menzies KJ, Ryu D, Wegner CJ, Wang X, Ropelle ER, Moullan N, Zhang H, Perino A, Lemos V, et al. Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology;63:1190–1204 (2016); DOI: https://doi.org/10.1002/hep.28245.
[48] Trammell SA, Schmidt MS, Weidemann BJ, Redpath P, Jaksch F, Dellinger RW, Li Z, Abel ED, Migaud ME, Brenner C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat Commun;7: 12948 (2016); DOI: https://doi.org/10.1038/ncomms12948.
[49] Airhart SE, Shireman LM, Risler LJ, Anderson GD, Naganda Gowda GA, Raftery D, Tian R, Shen DD, O’Brien KD. An open-label, non-randomized study of the pharmacokinetics of the nutritional supplement nicotinamide riboside (NR) and its effects on blood NAD+ levels in healthy volunteers. PLOS ONE (2017); DOI: https://doi.org/10.1371/journal.pone.0186459.
[50] Heilbronn LK. Clinical Trials Corner. Nutr Healthy Aging; 4:193–194 (2017); DOI: https://doi.org/ 10.3233/NHA-170001.
[51] Tsilioni I, Taliou A, Francis K, Theoharides TC. Children with autism spectrum disorders, who improved with a luteolin-containing dietary formulation, show reduced serum levels of TNF and IL-6. Transl Psychiatry; 5:e647 (2015); DOI: https://doi.org/10.1038/tp.2015.142.
[52] Essuman K, Summers DW, Sasaki Y, Mao X, DiAntonio A, Milbrandt J. The SARM1 Toll/Interleukin-1 Receptor Domain Possesses Intrinsic NAD+ Cleavage Activity that Promotes Pathological Axonal Degeneration. Neuron; 93:1334–1343 e1335 (2017); DOI: https://doi.org/10.1016/j.neuron.2017.02.022.
[53] Cerutti R, Pirinen E, Lamperti C, Marchet S, Sauve AA, Li W, Leoni V, Schon EA, Dantzer F, Auwerx J, et al. NAD(+)-dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metab; 19:1042–1049 (2014); DOI: https://doi.org/10.1016/j.cmet.2014.04.001.
[54] Ross Grant, Jade Berg, Richard Mestayer, Nady Braidy, James Bennett, Susan Broom, and James Watson, A Pilot Study Investigating Changes in the Human Plasma and Urine NAD+ Metabolome During a 6 Hour Intravenous Infusion of NAD+, Front Aging Neurosci. 11: 257 (2019), DOI: https://doi.org/10.3389/fnagi.2019.00257.
How to cite and Reference