


Hemoglobinopathies like sickle cell disease (SCD) and beta thalassemia are the most common monogenic disorders and the number of children born every year with such conditions is predicted to reach about 400,000 by the year 2050. Transplantation of haematopoietic stem cells remains the gold standard of treatment but finding a Human Leukocyte Antigen (HLA)-matched donors is a major bottle neck when large numbers of patients need to be treated. Recent advances in gene editing tools, like Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR), show immense potential to transform the gene/cell therapy scenario. This review covers recent breakthroughs in treating SCD and transfusion-dependent beta thalassemia such as re-initiation of fetal hemoglobin expression, and the planned clinical trials to correct the sickle cell mutation through CRISPR-based gene repair strategies. Further, expression of Factor IX to treat hemophilia using zinc finger nucleases, and universal Chimeric Antigen Receptor T cells (CAR-T) to treat cancers using genome editing are also discussed. Finally, challenges in developing these gene editing tools for clinical translation, along with ensuring their safety, feasibility, and affordability, are also addressed.
*Corresponding Author
E-mail: gokul.kesavan@vowelslifesciences.com (Gokul Kesavan)
Keywords: Genome editing, CRISPR Therapy, Haemoglobinopathies, Mutagenesis, Gene Therapy
Introduction
Worldwide, about 300,000 infants are born with Sickle cell anaemia every year and most of these births happen in sub-Saharan Africa, the Middle East, and in India1. Sickle cell disease (SCD) is a generic term that encompasses a group of inherited conditions such as sickle cell anaemia, HbSC (a milder sickling disease with one Haemoglobin S and Haemoglobin C allele)- and b-thalassemia, which are disorders characterized by mutations in the haemoglobin b-subunit (HbB). A missense mutation in the b-globin gene, referred to as the sickle haemoglobin (HbS) allele, results in disease phenotype when homozygous. Described more than 100 years ago, SCD is the first genetic disorder where effective therapy has remained elusive even though the causative mutation has been identified2,3; nevertheless, currently available treatments include administration of hydroxycarbamide (hydroxyurea), erythrocyte transfusion, or haemopoietic stem cell transplantation. SCD phenotypes include haemolytic anaemia, and it ultimately leads to multisystem disorder that affects almost all organs in the human body due to vaso-occlusion or blockage of small blood vessels (for extensive review on SCD please see4,5).
Haemoglobin (Hb) is a tetrameric protein that is formed from different combinations of globin subunits and an oxygen-carrying haem moiety nestled among them. The most abundant form of haemoglobin present in adults is HbA, which contains two alpha chains (encoded by HBA1 and HBA2 genes) and two beta chains (encoded by HbB genes), hence it is designated a2b2. In contrast, foetal haemoglobin (HbF) contains two alpha and two gamma chains (a2g2). The switch from HbF to HbA occurs in the early postnatal months and is referred to as the “foetal-to-adult haemoglobin switch”. In adults, just about 1% of total haemoglobin is HbF. Transcription factors are involved in the switch from HbF to HbA and BCL11A is one such factor that repress postnatal HbF expression; thus, neonates and infants with b-thalassemia or SCD are asymptomatic as long as HbF levels remain high but turn symptomatic during their first year of life when HbF levels decline and HbA levels increase. Interestingly, persistent expression of HbF into adulthood due to hereditary causes in patients with SCD or b-thalassemia results in little or no disease6. Consistently, deletion of the erythroid-specific enhancer, BCL11A, in a transgenic mice model of SCD, corrects the haematological and pathological phenotypes associated with SCD and b-thalassemia through enhanced expression of HbF6,7.
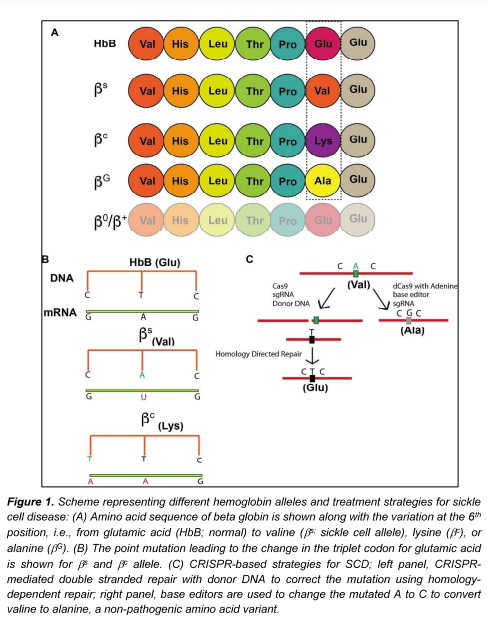
Sickle haemoglobin (HbS) is formed due to a point mutation in the HbB gene encoding the sixth amino acid due to which glutamic acid is replaced by valine, and the mutated allele is donated as bs. SCD occurs when both the HbB alleles are mutated and at least one of them is bs. In case of bc, the glutamic acid is replaced by lysine, which typically leads to milder episodes of haemolytic anaemia and fewer complications compared to patients with homozygous bs alleles. In individuals with a non-pathogenic variant called the “Makassar variant”, first identified in Makassar, Sulawesi, Republic of Indonesia, glutamic acid is switched to alanine8 and the allele is represented as bG. Individuals with a single bs allele i.e., HbB/bs have the sickle cell trait but not SCD (Fig 1A, 1B). Mutations in HbB that cause transfusion dependent b-thalassemia (TDT) result in reduced (denoted as b+ allele) or absent b globin synthesis (denoted as b0 allele). All disease-causing mutant alleles of the b-globin are depicted in Fig 1A.
This review aims to discuss the tremendous progress that has been made in the last five years in cutting edge gene editing strategies such as CRISPR and zinc finger nuclease for treating various hemoglobinopathies, especially common blood disorders like sickle cell disease, beta thalassemia and haemophilia. Next, potential treatment methods, including manipulation of upstream regulators or correcting mutant genes, provide options for permanently curing the disease in millions of affected individuals, which is in contrast to existing treatment procedures that only address the disease phenotype. The final section deals with how CRISPR-edited allogenic “off-the-shelf” cells can be used to address certain obstacles associated with immunotherapies for hematopoietic cancers like leukaemia’s and lymphomas.
Emerging Therapies for SCD and b- Thalassemia
Gene editing offers the ability to rewrite the genetic code and thereby correct mutations responsible for various inherited genetic disorders. Currently, the most popular genome editing tool is CRISPR, which was originally identified as a bacterial immune system that can recognize and eliminate DNA from invading or foreign bacteriophages9. Thus, CRISPR with its nuclease Cas, is referred to as the CRISPR/Cas system and it has been shown to potentially revolutionize agriculture, biotechnology, and modern medicine10. For example, the CRISPR nuclease Cas9 is directed by a short guide RNA that is complementary to target DNA, which results in the formation of a DNA-RNA complex. The Cas9 protein has two nuclease domains, namely the Ruvc and HNH domain, that cause double-stranded breaks at the target site (extensively reviewed in10,11). These double-stranded breaks are repaired through two mechanisms, namely, either the error prone Non-Homologous End Joining (NHEJ) method, which causes insertions or deletions at the cut site (referred as indels), or the less frequent but more efficient Homology Directed Repair (HDR). The error prone NHEJ mechanism is commonly used to create frame shift or deletion mutations to study gene function while the HDR strategy is used for correcting mutations in the genome using donor DNA. An exhaustive list of clinical trials on treating various blood disorders that involve gene editing are listed in Table 1.
In a pioneering study, two patients, one with SCD and other with TDT, were treated with CRISPR-edited hematopoietic stem and progenitor cells (HSPC, CD34+), referred to as CDX001 (NCT03655678 and NCT0345287)12, wherein previously, CRISPR/Cas9-mediated saturating mutagenesis of non-coding elements at the BCL11A locus was used to identify an erythroid-specific targeting sequence13. Mutation at this erythroid-specific enhancer locus resulted in the disruption of GATA1 transcription factor binding and consequent BCL11A repression, but only in the erythroid lineage. Importantly, this mutation evaded neurological and immunological adverse effects observed in conventional knockouts, thereby supporting its potential for clinical applications12. Silencing this erythroid-specific enhancer (BCL11A) enabled the CRISPR-mediated gene edited HSPCs to reinitiate production of foetal haemoglobin (HbF) and reactivate g-globin expression, consequently improving disease phenotype. Importantly, 8 more patients (6 with TDT and 2 with SCD), also treated with CTX001, showed results comparable to those seen in the first 2 patients during a 3-month follow up period, attesting to the feasibility of this therapeutic strategy12. Interestingly, two previous clinical trials (NCT03432364, NCT03653247) had utilized Zinc Finger Nuclease (ZFN)-based gene editing to silence the BCL11A gene to restore HbF expression and the results are yet to be published. A similar study published concurrently used lentivirus-mediated expression of short hairpin RNA (shRNA) for post-transcriptional gene silencing of BCL11A (BCH-BB694) and showed restoration of HbF expression in SCD patients. All the six patients in this study showed a reduction in or absence of clinical manifestations associated with SCD during the follow up period of about 18 months14. Notably, both the CRISPR and ShRNA strategies involved isolation of HSPCs and their ex-vivo manipulation prior to infusion of the modified cells into patients; thus, both approaches require patients to undergo harsh chemotherapy to eliminate defective stem cells and permit marrow repopulation by the modified cells.
A phase1/2 clinical trial (NCT04774536) at the University of California at Berkeley and at Los Angeles plans to use CRISPR-edited HSPCs (CRISPR_SCD001) that will be transplanted in patients suffering from severe SCD with the aim of correcting the faulty beta-globin gene ex vivo; this represents the first gene correction therapy for SCD. It must be pointed out that the CDX001 and zinc finger nuclease approaches enhanced HbF expression to compensate for the defective sickle Hb rather than correct the sickle cell mutation, as with SCD001. Similarly, Graphite bio, a US biotech company has obtained an Investigational New Drug (IND) approval to initiate clinical trials for GPH101 to treat SCD (NCT04819841). Here, GPH101 relies on the HDR mechanism to cut out the disease-causing mutation and replace it with the correct sequence using a repair template (Fig. 1C). Graphite bio is also aiming to use this gene correction strategy to treat other diseases like XSCID, a life-threatening immune disease, and to express glucocerebrosidase in a safe harbour site (at the CCR5 locus) to treat various types of Gaucher’s disease. Both CRISPR_SCD001 and GPH101 are ex vivo CRISPR approaches that require isolation and culturing of cells in vitro for gene editing.
In contrast to the above, base editors are CRISPR-based gene editing tools that do not create a double-stranded break, instead, they chemically modify the nucleotide base at the target sequence, and thereby reduce the incidence of insertion or deletion mutations. In a recent study using animal models and human cells, the sickle cell mutation, bs, was corrected to a non-pathogenetic variant bG (also called Makassar variant) using the adenosine base editor (ABE; Fig. 1C)15. Beam therapeutics, a USA-based biotechnology company, has plans to take this base editing strategy to clinical trials to treat several other diseases that are associated with point mutations.
Haemophilia and gene therapy with Zinc Finger Nuclease
Haemophilia A and B are the most common forms of bleeding disorders and are due to the deficiency of factors FVIII and FIX, respectively. Administration of plasma or factor concentrates, 2 to 3 times per week, is required due to the short half-life of plasma proteins, which increases both treatment burden and cost. About 70% of all haemophilia is type A, which is an X-linked disorder that only affects males; females are typically only carriers. Recently, bioengineered factors with decreased immunogenicity, increased efficiency, and extended half-life have been developed, which are easy to administer. Further, administration of adeno associated virus (AAV), containing a synthetic liver-specific promoter (LPI-hFIXco) that drives the transgenic expression of FIX, has been shown to have stable expression over a 7-year follow up period (NCT00979238), which resulted in a substantial reduction in spontaneous bleeding and FIX protein usage without toxicity. However, the AAV genome is maintained in episomal format i.e remains independent and not integrating with the chromosomes, which could lead to potential loss of transgene expression when the transduced cells undergo continuous cell division16.
Zinc finger nuclease (ZFN) are genome editors that consist of a zinc finger DNA binding domain that specifies nuclease activity using the Fok1 endonuclease. A pair of ZFNs are designed for a target site and dimerization of the Fok1 domain creates double-stranded breaks17. Sangamo therapeutics, a gene therapy company, has designed a ZFN-based transgenic approach to treat Hemophilia B by expressing FIX. ZFN-mediated double stranded breaks were created to insert the correct copy of FIX under the albumin gene promoter and appropriate regulatory elements to ensure strong FIX expression. As intravenous delivery of the ZFN complex as AAV vectors will ensure their delivery to predominantly hepatocytes, the transgene will be inserted and expressed in the liver. A clinical trial (NCT02695160) was initiated in 2016 but is currently terminated and no results are available. Several preclinical studies have used CRISPR/Cas tools to successfully express human FIX in animal models or to correct mutations in induced pluripotent stem cells18,19, and it is highly possible that these promising studies will progress to clinical trials.
CRISPR-based therapies for blood-associated cancers
Cancer is one of the leading causes of death and the heterogeneity of cancer cells in a patient renders treatment challenging. New therapeutic strategies involving immunotherapy show promising results and are gaining popularity due to the close relationship between cancer cells and the immune system. Cancer is considered to progress through the following stages. In the first stage, called the “elimination” stage, both innate and adaptive immunity recognize and destroy developing tumors, with these processes occurring before the development of any clinical symptoms. The second stage, called the “equilibrium” stage, is the longest stage where the immune system continues to eliminate cancer cells, and when this fails, cancer advances to the “escape” stage, wherein cancer cells are conferred resistance to immune detection and tumors become clinically detectable20.
Chimeric Antigen Receptors (CAR-T) cells are engineered immune cells that can seek and destroy cancer cells in different stages of the disease and the FDA (USA) approved two such CAR-T therapies in 2017. Autologous CAR-T therapy is a commonly used strategy where the patient’s T-cells are genetically modified. This process is time consuming as the manufacturing and quality control steps need to be carried for every individual patient, and consequently, raise production cost. However, allogenic CAR-T cells offer a promising strategy to provide “off-the shelf” cells that can be manufactured in bulk, thereby dramatically reducing both production cost and time.
CRISPR-mediated gene knockouts in immune cells aim to avoid graft rejection and facilitate their use as universal cells that can be not only used to treat large numbers of patients but also insert the CAR gene21. Specifically, T-cell receptor genes (TCR) and β2-microglobulin (β2M), which are essential for presenting Human Leukocyte Antigen type 1 (HLA1) molecules, are knocked out using CRISPR/Cas-based editing, and as these allogenic universal cells can also be prepared from healthy donors, several biotechnology companies, like CRISPR therapeutics, are manufacturing such CAR-T cells that are less likely to be rejected by host cells22. Multiple clinical trials are currently underway to test the safety, feasibility, and efficiency of allogenic CAR-T cells in eliminating leukemia, lymphoma, and B-cell malignancies (NCT03166878, NCT03398967 and NCT04035434). Universal CAR-T cells also reduce the dependance on the patient’s own T-cells, which may not be present in sufficient numbers.
Table1: Clinical trials for blood disorders that use genome editing tools
|
Clinical trial NO |
Disease | Strategy | Status (July 2021) |
| NCT03432364
|
Beta Thalassemia | ZFN mediated BCL11A mutation | Active, not recruiting |
| NCT03653247 | Sickle cell disease | ZFN mediated BCL11A mutation | Recruiting |
| NCT03655678 | Transfusion dependent beta thalassemia | CRISPR/Cas9 mediated BCL11A mutation | Recruiting |
| NCT03745287 | Sickle cell disease | CRISPR/Cas9 mediated BCL11A mutation | Recruiting |
| NCT04774536 | Sickle cell disease | CRISPR/Cas9 mediated gene correction in CD34+ cells | Not yet Recruiting |
| NCT04819841 | Sickle cell disease | CRISPR/Cas9 mediated gene correction in CD34+ cells | Not yet Recruiting |
| NCT03728322 | Beta Thalassemia | CRISPR/Cas9 mediated gene correction in iHSC cells | Unknown |
| NNCT02695160
|
Haemophilia B | ZFN mediated transgenic expression of FIX | Terminated |
| NCT03166878 | Leukaemia and Lymphoma | CD-19 directed CAR-T cells CRISPR edited to disrupt TCR and B2M genes to create universal CAR-T cells | Recruiting |
| NCT03398967 | Leukaemia and Lymphoma | Dual specificity CD-19, CD-20 or CD-22 directed universal CAR-T cells | Recruiting |
| NCT04035434 | B- cell malignancies | CD-19 CAR-T cells, CRISPR mediated editing to make these cells allogenic | Recruiting |
Conclusions and perspectives
Meganucleases such as I-Sec1, ZFNs, and transcription activator-like effector (TALEN) can generate double stranded breaks and complete homology-directed repair. However, difficulties in cloning and protein engineering for ZFN and TALENs, along with difficulties in finding a meganuclease target site at the desired locus, have curtailed broad application of these genome editors. Alternatively, CRISPR offers robust and high efficiency editing with simpler and easy-to-use reagent preparations; these attributes make it one of the most utilized genome editing tools, both in the laboratory and in translation to clinical trials. This is reflected in upcoming or planned clinical trials to correct SCD mutations and implement allogenic CAR-T based cell therapies that use CRISPR. One of the major challenges of such genome editing tools, including CRISPR, is off-target effects (OFE) i.e., nucleases cutting at unintended locations due to target sequence homology, but multiple strategies, including the use of high-fidelity variants of Cas9, like xCas9, and other Cas proteins with minimal OFE (like Cpf1) have been recently shown to perform better in preclinical studies11. Additionally, improved in silico methods to optimize target design, followed by whole genome sequencing approaches to detect off targets in vivo, are being used to rigorously test and eliminate OFE prior to clinical trials. A promising alternative strategy is the use of base editors to correct point mutations to reduce off target insertions and/or deletions as base editing tools do not cut the DNA23.
CRISPR-based gene correction strategies for SCD utilize electroporation techniques to deliver components to stem cells in vitro and predictions suggest that gene correction in even 20% of the stem cells would be sufficient to out compete the native sickle cells and result in strong clinical benefits. The above-mentioned strategy is an example of ex vivo CRISPR therapy that involves autologous stem cell isolation, expansion, and gene modification in vitro, followed by validation for on- and off-target effects, and finally, reinfusion of the corrected cells into patients; thus, the need for multiple steps will obviously increase time and cost of treatment. Therefore, in vivo CRISPR therapies will gain popularity as they avoid cell isolation and in vitro manipulation of these isolated cells. However, currently, in vivo therapies are limited to few organs like the retina or liver as delivery of gene editing components to these sites is relatively easy24,25. Alternatively, allogenic universal donor cells, as in the clinical trials for CAR-T therapies, are an efficient process for mass producing transplantable cells, but the stability and efficiency of such cells in treating diseases are yet to be established26. Treatment cost is a major factor that will determine the wide-spread use of such therapies for blood disorders like SCD and TDT in low-income countries. For example, in India, bone marrow-derived stem cells are the major cell source for therapy and combining gene editing with existing stem cell therapies can be used to create a platform that can treat multiple blood disorders27. Nonetheless, how these groundbreaking innovations can be made available and affordable to large numbers of patients across the globe remains to be seen.
Acknowledgements
The author is thankful to Dr. Vasuprada Iyengar for language and content editing. The author acknowledges that the number of published papers, both experimental and reviews, far exceed those cited here. The author aimed to highlight only the most current strategies that show potential for wide application to large populations, and any omission reflects this aim alone.
Funding
No funding was received to assist the preparation of this manuscript. The author declares no financial interest; G.K is employed at Vowels Lifesciences Private Limited, a healthcare company associated with cord blood banking and teaching activities.
Conflict of interest
The author declares no conflict of interest.
Reference:
(1) Piel, F. B.; Hay, S. I.; Gupta, S.; Weatherall, D. J.; Williams, T. N. Global Burden of Sickle Cell Anaemia in Children under Five, 2010–2050: Modelling Based on Demographics, Excess Mortality, and Interventions. PLoS Med. 2013, 10 (7), e1001484. https://doi.org/10.1371/journal.pmed.1001484.
(2) HERRICK, J. B. PECULIAR ELONGATED AND SICKLE-SHAPED RED BLOOD CORPUSCLES IN A CASE OF SEVERE ANEMIA. Arch. Intern. Med. 1910, VI (5), 517–521. https://doi.org/10.1001/archinte.1910.00050330050003.
(3) Ingram, V. M. A Specific Chemical Difference Between the Globins of Normal Human and Sickle-Cell Anæmia Hæmoglobin. Nature 1956, 178 (4537), 792–794. https://doi.org/10.1038/178792a0.
(4) Kato, G. J.; Piel, F. B.; Reid, C. D.; Gaston, M. H.; Ohene-Frempong, K.; Krishnamurti, L.; Smith, W. R.; Panepinto, J. A.; Weatherall, D. J.; Costa, F. F.; Vichinsky, E. P. Sickle Cell Disease. Nat. Rev. Dis. Primer 2018, 4 (1), 1–22. https://doi.org/10.1038/nrdp.2018.10.
(5) Piel, F. B.; Steinberg, M. H.; Rees, D. C. Sickle Cell Disease https://www.nejm.org/doi/pdf/10.1056/NEJMra1510865 (accessed 2021 -07 -01). https://doi.org/10.1056/NEJMra1510865.
(6) Bauer, D. E.; Kamran, S. C.; Lessard, S.; Xu, J.; Fujiwara, Y.; Lin, C.; Shao, Z.; Canver, M. C.; Smith, E. C.; Pinello, L.; Sabo, P. J.; Vierstra, J.; Voit, R. A.; Yuan, G.-C.; Porteus, M. H.; Stamatoyannopoulos, J. A.; Lettre, G.; Orkin, S. H. An Erythroid Enhancer of BCL11A Subject to Genetic Variation Determines Fetal Hemoglobin Level. Science 2013, 342 (6155), 253–257. https://doi.org/10.1126/science.1242088.
(7) Xu, J.; Peng, C.; Sankaran, V. G.; Shao, Z.; Esrick, E. B.; Chong, B. G.; Ippolito, G. C.; Fujiwara, Y.; Ebert, B. L.; Tucker, P. W.; Orkin, S. H. Correction of Sickle Cell Disease in Adult Mice by Interference with Fetal Hemoglobin Silencing. Science 2011, 334 (6058), 993–996. https://doi.org/10.1126/science.1211053.
(8) Viprakasit, V.; Wiriyasateinkul, A.; Sattayasevana, B.; Miles, K. L.; Laosombat, V. Hb G-Makassar [Beta6(A3)Glu–>Ala; Codon 6 (GAG–>GCG)]: Molecular Characterization, Clinical, and Hematological Effects. Hemoglobin 2002, 26 (3), 245–253. https://doi.org/10.1081/hem-120015028.
(9) Mojica, F. J.; Díez-Villaseñor, C.; Soria, E.; Juez, G. Biological Significance of a Family of Regularly Spaced Repeats in the Genomes of Archaea, Bacteria and Mitochondria. Mol. Microbiol. 2000, 36 (1), 244–246. https://doi.org/10.1046/j.1365-2958.2000.01838.x.
(10) Hsu, P. D.; Lander, E. S.; Zhang, F. Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell 2014, 157 (6), 1262–1278. https://doi.org/10.1016/j.cell.2014.05.010.
(11) Pickar-Oliver, A.; Gersbach, C. A. The next Generation of CRISPR–Cas Technologies and Applications. Nat. Rev. Mol. Cell Biol. 2019, 20 (8), 490–507. https://doi.org/10.1038/s41580-019-0131-5.
(12) Frangoul, H.; Altshuler, D.; Cappellini, M. D.; Chen, Y.-S.; Domm, J.; Eustace, B. K.; Foell, J.; de la Fuente, J.; Grupp, S.; Handgretinger, R.; Ho, T. W.; Kattamis, A.; Kernytsky, A.; Lekstrom-Himes, J.; Li, A. M.; Locatelli, F.; Mapara, M. Y.; de Montalembert, M.; Rondelli, D.; Sharma, A.; Sheth, S.; Soni, S.; Steinberg, M. H.; Wall, D.; Yen, A.; Corbacioglu, S. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N. Engl. J. Med. 2021, 384 (3), 252–260. https://doi.org/10.1056/NEJMoa2031054.
(13) Canver, M. C.; Smith, E. C.; Sher, F.; Pinello, L.; Sanjana, N. E.; Shalem, O.; Chen, D. D.; Schupp, P. G.; Vinjamur, D. S.; Garcia, S. P.; Luc, S.; Kurita, R.; Nakamura, Y.; Fujiwara, Y.; Maeda, T.; Yuan, G.-C.; Zhang, F.; Orkin, S. H.; Bauer, D. E. BCL11A Enhancer Dissection by Cas9-Mediated in Situ Saturating Mutagenesis. Nature 2015, 527 (7577), 192–197. https://doi.org/10.1038/nature15521.
(14) Esrick, E. B.; Lehmann, L. E.; Biffi, A.; Achebe, M.; Brendel, C.; Ciuculescu, M. F.; Daley, H.; MacKinnon, B.; Morris, E.; Federico, A.; Abriss, D.; Boardman, K.; Khelladi, R.; Shaw, K.; Negre, H.; Negre, O.; Nikiforow, S.; Ritz, J.; Pai, S.-Y.; London, W. B.; Dansereau, C.; Heeney, M. M.; Armant, M.; Manis, J. P.; Williams, D. A. Post-Transcriptional Genetic Silencing of BCL11A to Treat Sickle Cell Disease. N. Engl. J. Med. 2021, 384 (3), 205–215. https://doi.org/10.1056/NEJMoa2029392.
(15) Newby, G. A.; Yen, J. S.; Woodard, K. J.; Mayuranathan, T.; Lazzarotto, C. R.; Li, Y.; Sheppard-Tillman, H.; Porter, S. N.; Yao, Y.; Mayberry, K.; Everette, K. A.; Jang, Y.; Podracky, C. J.; Thaman, E.; Lechauve, C.; Sharma, A.; Henderson, J. M.; Richter, M. F.; Zhao, K. T.; Miller, S. M.; Wang, T.; Koblan, L. W.; McCaffrey, A. P.; Tisdale, J. F.; Kalfa, T. A.; Pruett-Miller, S. M.; Tsai, S. Q.; Weiss, M. J.; Liu, D. R. Base Editing of Haematopoietic Stem Cells Rescues Sickle Cell Disease in Mice. Nature 2021, 1–8. https://doi.org/10.1038/s41586-021-03609-w.
(16) Nathwani, A. C. Gene Therapy for Hemophilia. Hematol. Am. Soc. Hematol. Educ. Program 2019, 2019 (1), 1–8. https://doi.org/10.1182/hematology.2019000007.
(17) Urnov, F. D.; Rebar, E. J.; Holmes, M. C.; Zhang, H. S.; Gregory, P. D. Genome Editing with Engineered Zinc Finger Nucleases. Nat. Rev. Genet. 2010, 11 (9), 636–646. https://doi.org/10.1038/nrg2842.
(18) Morishige, S.; Mizuno, S.; Ozawa, H.; Nakamura, T.; Mazahery, A.; Nomura, K.; Seki, R.; Mouri, F.; Osaki, K.; Yamamura, K.; Okamura, T.; Nagafuji, K. CRISPR/Cas9-Mediated Gene Correction in Hemophilia B Patient-Derived IPSCs. Int. J. Hematol. 2020, 111 (2), 225–233. https://doi.org/10.1007/s12185-019-02765-0.
(19) Wang, L.; Yang, Y.; Breton, C. A.; White, J.; Zhang, J.; Che, Y.; Saveliev, A.; McMenamin, D.; He, Z.; Latshaw, C.; Li, M.; Wilson, J. M. CRISPR/Cas9-Mediated in Vivo Gene Targeting Corrects Hemostasis in Newborn and Adult Factor IX–Knockout Mice. Blood 2019, 133 (26), 2745–2752. https://doi.org/10.1182/blood.2019000790.
(20) Miliotou, A. N.; Papadopoulou, L. C. CAR T-Cell Therapy: A New Era in Cancer Immunotherapy. Curr. Pharm. Biotechnol. 2018, 19 (1), 5–18. https://doi.org/10.2174/1389201019666180418095526.
(21) Stadtmauer, E. A.; Fraietta, J. A.; Davis, M. M.; Cohen, A. D.; Weber, K. L.; Lancaster, E.; Mangan, P. A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; Tian, L.; Gonzalez, V. E.; Xu, J.; Jung, I.; Melenhorst, J. J.; Plesa, G.; Shea, J.; Matlawski, T.; Cervini, A.; Gaymon, A. L.; Desjardins, S.; Lamontagne, A.; Salas-Mckee, J.; Fesnak, A.; Siegel, D. L.; Levine, B. L.; Jadlowsky, J. K.; Young, R. M.; Chew, A.; Hwang, W.-T.; Hexner, E. O.; Carreno, B. M.; Nobles, C. L.; Bushman, F. D.; Parker, K. R.; Qi, Y.; Satpathy, A. T.; Chang, H. Y.; Zhao, Y.; Lacey, S. F.; June, C. H. CRISPR-Engineered T Cells in Patients with Refractory Cancer. Science 2020, 367 (6481). https://doi.org/10.1126/science.aba7365.
(22) Salas-Mckee, J.; Kong, W.; Gladney, W. L.; Jadlowsky, J. K.; Plesa, G.; Davis, M. M.; Fraietta, J. A. CRISPR/Cas9-Based Genome Editing in the Era of CAR T Cell Immunotherapy. Hum. Vaccines Immunother. 2019, 15 (5), 1126–1132. https://doi.org/10.1080/21645515.2019.1571893.
(23) Porto, E. M.; Komor, A. C.; Slaymaker, I. M.; Yeo, G. W. Base Editing: Advances and Therapeutic Opportunities. Nat. Rev. Drug Discov. 2020, 19 (12), 839–859. https://doi.org/10.1038/s41573-020-0084-6.
(24) Gillmore, J. D.; Gane, E.; Taubel, J.; Kao, J.; Fontana, M.; Maitland, M. L.; Seitzer, J.; O’Connell, D.; Walsh, K. R.; Wood, K.; Phillips, J.; Xu, Y.; Amaral, A.; Boyd, A. P.; Cehelsky, J. E.; McKee, M. D.; Schiermeier, A.; Harari, O.; Murphy, A.; Kyratsous, C. A.; Zambrowicz, B.; Soltys, R.; Gutstein, D. E.; Leonard, J.; Sepp-Lorenzino, L.; Lebwohl, D. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N. Engl. J. Med. 2021, 0 (0), null. https://doi.org/10.1056/NEJMoa2107454.
(25) Maeder, M. L.; Stefanidakis, M.; Wilson, C. J.; Baral, R.; Barrera, L. A.; Bounoutas, G. S.; Bumcrot, D.; Chao, H.; Ciulla, D. M.; DaSilva, J. A.; Dass, A.; Dhanapal, V.; Fennell, T. J.; Friedland, A. E.; Giannoukos, G.; Gloskowski, S. W.; Glucksmann, A.; Gotta, G. M.; Jayaram, H.; Haskett, S. J.; Hopkins, B.; Horng, J. E.; Joshi, S.; Marco, E.; Mepani, R.; Reyon, D.; Ta, T.; Tabbaa, D. G.; Samuelsson, S. J.; Shen, S.; Skor, M. N.; Stetkiewicz, P.; Wang, T.; Yudkoff, C.; Myer, V. E.; Albright, C. F.; Jiang, H. Development of a Gene-Editing Approach to Restore Vision Loss in Leber Congenital Amaurosis Type 10. Nat. Med. 2019, 25 (2), 229–233. https://doi.org/10.1038/s41591-018-0327-9.
(26) Meissner, T. B.; Cowan, C. A. More Bang for Your Buck: “Off-The-Shelf” Solutions for Cell Replacement Therapy. StemJournal 2020, 2 (1), 1–5. https://doi.org/10.3233/STJ-200002.
(27) Boopalan, D.; Pandian, R.; Kesavan, G. Prospects for Stem Cell-Based Regenerative Therapies in India. StemJournal 2021, 3 (1), 11–21. https://doi.org/10.3233/STJ-210002.
/div